
Federal law prohibits the distribution of new drugs or biologics until the FDA has reviewed clinical data and determined that a particular product is safe and effective for a specific use in human subjects.
In order to test a new drug or biologic in clinical trials, it is necessary to obtain an exemption from this law. Thus a drug sponsor is required to apply for an Investigational New Drug (IND) exemption before tests with human subjects may begin.
In general, the review requirements for biologics are the same as those for drugs. Accordingly, unless otherwise indicated, the provisions that follow use of the term "drug," apply to biologics as well as to drugs. The investigator is responsible for obtaining the IND number and providing it to the IRB. Studies that involve FDA-regulated products that are submitted without a IND number will be reviewed by the IRB with respect to determining the need for an IND , based on federal requirements and the investigator's response to questions contained in the protocol.
If the IRB determines that the study does not require an IND and approves the study, the study may begin. If the IRB determines that an IND is needed, the investigator/sponsor must submit an IND application to the FDA and provide documentation of the outcome of the FDA determination ( IND number) to the IRB before the IRB gives approval to enroll subjects in the study.
An IND is an application to the FDA for permission to test a drug to determine if it is safe and effective. The process is governed by 21CFR 312.
The IRB may consider a study using a drug product that is lawfully marketed in the United States to be exempt from the requirements for obtaining an IND if all the following apply:
- The investigation is not intended to be reported to FDA as a well-controlled study in support of a new indication for use nor intended to be used to support any other significant change in the labeling for the drug;
- If the drug that is undergoing investigation is lawfully marketed as a prescription drug product, the investigation is not intended to support a significant change in the advertising for the product;
- The investigation does not involve a route of administration or dosage level or use in a subject population (e.g., children, prisoners, pregnant women and fetuses) or other factor that significantly increases the risks (or decreases the acceptability of the risks) associated with the use of the drug product;
- The investigation is conducted in compliance with the requirements for institutional review and with the requirements for informed consent; and
- The investigation is conducted in compliance with the requirements with regard to promotion
- If a newly developed drug/biologic that is not approved by the FDA and not licensed for marketing in the US is to be tested for safety and efficacy in one or more human subjects.
- If a drug/biologic previously approved by the FDA and licensed for marketing in the US is to be studied in one or more human subjects:
-
- with the intent to generate data leading to the approval of a new advertising claim. (For example the manufacture would like to be able to advertise that this new drug is as good as or better than an approved product.)
- for a new clinical indication (For example the drug may be approved for one clinical indication such as cognitive impairment in Alzheimer's, but there is a desire to see if those with cognitive impairment due to multiple sclerosis could also be helped.)
- in a population for which it was not previously approved.(For example if the product is approved when used in adults but there is a desire to use the product in minors.)
- if the drug/biologic is being given in an unapproved formulation, route or delivery system. (For example if the product is approved when given intravenously however there is a desire to be able give this product orally.)
- Unapproved combinations of approved concurrent therapies require an IND. (For example if there are 2 approved chemotherapeutic agents available for a certain diagnosis and there is a desire to see if better response and lower toxicities could be experienced if the products were used concurrently.)
- If a dietary supplement or botanical is being studied for its effect on disease in the proposed investigation (i.e., to cure, treat mitigate, prevent or diagnose disease including its associated symptoms, then it may be considered an investigational new drug and may be subject to IND requirements.
When is an IND not required (exempt from IND)?
An IND is not required if the drug/biologic under study is already licensed and approved by the FDA for marketing in the USA , if all of the following study conditions are met:
- The investigation is not intended to be reported to the FDA as a well-controlled study in support of a new indication for use nor intended to be used to support any significant change in the labeling for the drug/biologic
- The drug/biologic that is undergoing investigation is lawfully marketed as a prescription drug/biologic product and the investigation is not intended to support a significant change in the advertising for the product.
- The investigation does not involve a route of administration or dosage level or use in a patient population or other factors that significantly increases the risks (or decreases the acceptability of the risks) associated with the use of the drug/biologic product.
- The investigation is conducted in compliance with the requirement for institutional review set forth in 21CRF 56 and with the requirements for informed consent set forth in 21CFR 50.
- The investigation is conducted in compliance with the requirements of 21CRF 312.7 - meaning that the drug/biologic may not be represented as safe or effective for the purposes for which it is under investigation nor may it be commercially distributed or test marketed or sold.
If all of these conditions are met, then the study is considered IND exempt.
An IND is an application to the FDA for permission to test a drug to determine if it is safe and effective. The process is governed by 21CFR 312.
Who makes the determination regarding need for IND application?
The determination of whether or not an IND application is required is made by the IRB-HSR. The FDA may over-rule the IRB regarding the need for an IND. The School of Medicine Clinical Trials Office (SOM-CTO) staff is available to assist the investigator in contacting the FDA and writing a position statement to the IRB-HSR regarding the need for an IND. If it is determined that an IND is required, the SOM-CTO personnel are also available to advise researchers regarding the application preparation. In addition, the IRB requires researchers to obtain SOM- CTO approval for these types of studies. The SOM-CTO review includes careful attention to the Data Safety Monitoring Plan, and Data Collection Forms. This review must take place before the IRB will allow subjects to be enrolled.
The investigator is required to wait 30 days after submitting the IND application to the FDA before enrolling subjects. During this time the FDA scientists will review the materials submitted, and if necessary, request additional information or require modifications. The FDA may send the sponsor an IND#, however this is not an approval to proceed. The IRB will not provide approval to enroll subjects in the study until the 30-day time period has passed.
If a UVA faculty member is the principal investigator on an IND or IDE, the IRB-HSR will require an approval from the School of Medicine Clinical Trials Office prior to subjects enrolling in the protocol. Prior to granting approval, the SOM CTO will conduct of review of various items with special focus on areas of FDA interest such as- inclusion/exclusion criteria, safety plan , endpoints, data collection process and the communication plan with other sites , if multi-site. The staff of the SOM CTO will also review Sponsor responsibilities with the PI. Sponsor responsibilities for an IND are found at 21CFR312. Sponsor responsibilities for an IDE are found at 21CFR812.
Most INDs are passively approved. When the FDA receives the application, the FDA assigns an IND number. A letter is sent to the applicant providing the IND number, however, this number should not be mistaken as an "approval letter." In most cases, passive approval is assumed if there has been no formal contact from the FDA within 30 days of the submission of the IND application to the FDA. Studies cannot be initiated until after the 30-day period (and until the IRB has approved the study).
When the UVA PI has an approved IND , the PI is also referred to as the "holder of the IND." The PI takes on the responsibilities of sponsor as defined in the regulations. Once the 30-day waiting period is over and the study is approved by the IRB, the PI may initiate the research project.
Responsibilities of the UVA PI as IND holder are as follows:
- The PI is expected to update or file amendments to the IND with the FDA in a timely fashion when:
- the protocol and/or consent is/are amended in a manner that affects the safety of subjects, the scope of the investigation, or the study design.
- when an adverse event occurs that is considered serious, unexpected and related/possibly related, a report should be submitted to the FDA via telephone or fax within 7 days. A written report should be sent to the FDA within 7 days. Unanticipated problems must also be reported within 7 days.
- each time a new investigational site is added (if the study is multi-site) the PI/IND holder must submit the documentation for that site including the 1572 and current dated and signed CVs.
- each time a new investigator is added. An amended 1572 and current dated and signed CV will be anticipated by the FDA.
- the anniversary date of the IND is within 60 days of the original IND submission date. . An annual progress report to the FDA regarding research activity taking place under the IND should contain the following elements:
- a completed 1571 numbered sequentially
- title(s) of the protocol(s) operating under the IND with detailed enrollment information for each protocol (# enrolled to date, # entered since last report, # in study treatment, # in follow up, # completed, # withdrawals. Each of these numbers should be reported by totals and then by age group, gender and race.
- A summary for each protocol operating under this IND of the most frequent and most serious adverse events organized by body system for each protocol
- A summary for each protocol operating under this IND of all IND Safety reports for the last year.
- A list of all deaths noting the cause of death for each protocol operating under this IND.
- List of withdrawals noting the reason for withdrawal for each protocol operating under this IND.
Each update or amendment to the IND should contain:
-
- completed FDA Form 1571 numbered sequentially.
- Cover letter or narrative describing the purpose of the submission.
- Documentation for the amendment. (For example, if the protocol is being modified, then a copy of the protocol should be submitted. If a Serious Adverse Event is being reported then a FDA Form 3500A MedWatch or CIOMS should be submitted.)
- The PI is expected to keep all data secure.
- All data is expected to verifiable. This means that adequate source documentation and data collection forms are maintained. Please refer to the SOM-CTO and FDA websites for additional information. Links to both are available from the IRB-HSR website.
Expanded Access for Unapproved Drugs and Biologics
FDA information on Expanded Access to Investigational Drugs/Biologics
For Physicians: How to Request Single Patient Expanded Access
FDA's Expanded Access Contact Information
21 CFR 312 Subpart I.
Expanded Access to Investigational Drugs for Treatment Use – Questions and Answers.
Keywords, Definitions and Resources
Refer to the expanded access categories and Title 21 of the Code of Federal Regulations (21 CFR) for more detailed information about expanded access request types.
- 21 CFR 312.310 Individual patients, including emergency use
- 21 CFR 312.315 Intermediate-size patient populations
- 21 CFR 312.320 Treatment IND or treatment protocol
Under FDA regulations (21 CFR 312.300), expanded access is the use of an investigational drug/biologic (referred to as “test article”) outside of a clinical trial for the diagnosis, monitoring, or treatment of a serious disease or condition when there is no satisfactory alternative therapy to treat the patient’s disease or condition. In contrast, participants in clinical trials/studies are considered human subjects whether they are patients or healthy volunteers. The main distinction between expanded access and the use of an investigational drug in the clinical trials covered under an IND, is that in most cases, traditional clinical trials are for the purpose of collecting safety and effectiveness data. The purpose of expanded access is to provide treatment. While expanded access is not considered a clinical investigation, FDA submission and IRB review are required.
Criteria for all expanded access uses for drugs/biologics:
Under the applicable criteria in 21 CFR 312.310(a), the physician must determine that the probable risk to the person from the investigational drug/biologic is not greater than the probable risk from the disease or condition; and the FDA must determine that the patient cannot obtain the investigational drug/biologic under another IND or protocol.
312.305 Requirements for ALL expanded access use:
(1) Patient(s) have a serious or immediately life-threatening disease or condition, and there is no comparable or satisfactory alternative therapy to diagnose, monitor, or treat the disease or condition;
(2) The potential patient benefit justifies the potential risks of the treatment use and those potential risks are not unreasonable in the context of the disease or condition to be treated; and
(3) Providing the investigational drug for the requested use will not interfere with the initiation, conduct, or completion of clinical investigations that could support marketing approval of the expanded access use or otherwise compromise the potential development of the expanded access use.
3 types of expanded access for drugs/biologics:
Under FDA’s current regulations, there are three categories of expanded access as shown in the diagram that follows. The primary difference in the three types of access is the number of patient(s) participating.
- Expanded access for individual patients, includes (1) emergency use (21 CFR 312.310) and (2) non-emergency use
- Expanded access for intermediate-size patient populations (generally smaller than those typical of a treatment IND or treatment protocol — a treatment protocol is submitted as a protocol to an existing IND by the sponsor of the existing IND) (21 CFR 312.315)
- Expanded access for wide-spread treatment use through a treatment IND or treatment protocol (designed for use in larger patient populations) (21 CFR 312.320)
For each category of expanded access, there are two (2) types of regulatory submissions to the FDA to obtain the IND:
(1) An expanded access protocol submitted as a protocol amendment to an EXISTING IND (i.e., an expanded access protocol) or
(2) A NEW IND submission, which is separate and distinct from any existing INDs and is intended only to make a drug/biologic available for treatment use (i.e., an expanded access IND)
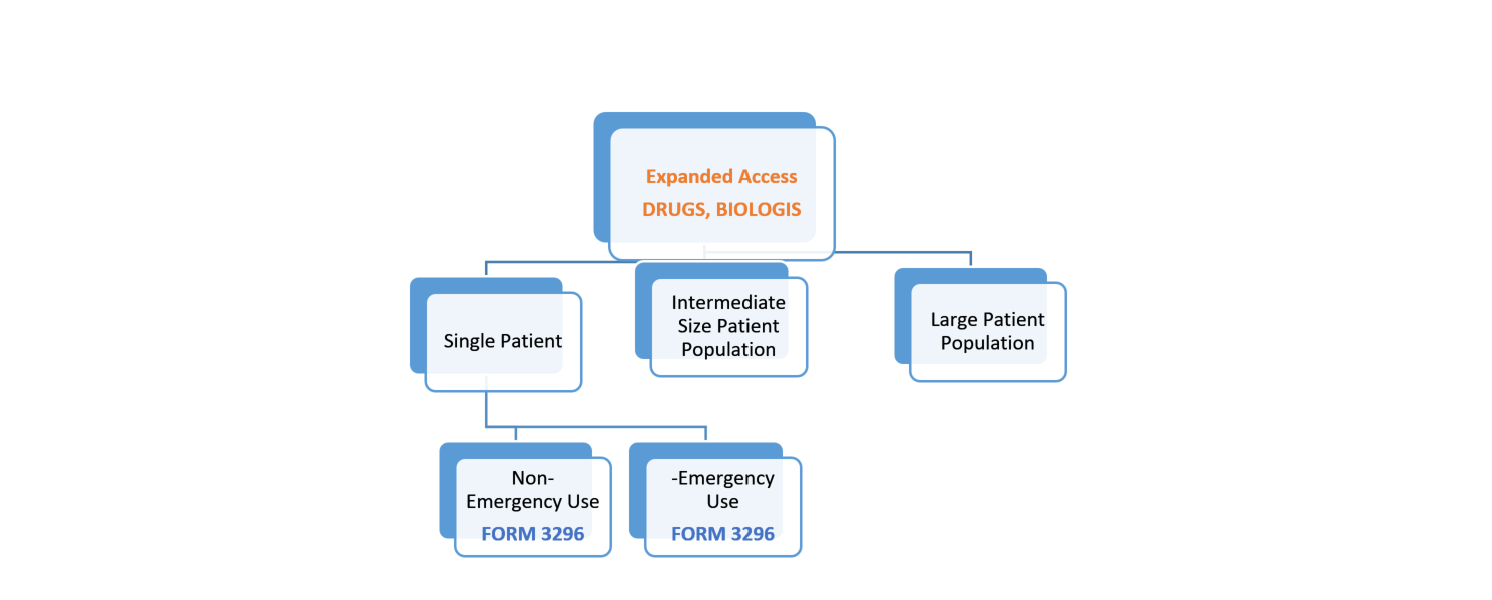
Single Patient Use [Emergency and Non-Emergency]:
For Emergency single patient treatment, you are NOT required to secure FDA and IRB review PRIOR to the use of the investigational treatment. Notification to the IRB must occur no later than 5 working days following administration of the test article.
For non-emergency single patient treatment, you ARE REQUIRED to notify the IRB PRIOR to the use of the test article and obtain FDA authorization before use.
Informed Consent: Informed consent must be sought from the subject or the subject’s legally authorized representative (LAR). The consent form should contain all the standard elements of informed consent. If the subject is unable to sign the consent, it may be signed by either a legal guardian or an attorney-in-fact with the authority to make health care decisions. If a person fulfilling those requirements is not available, review Virginia law allowing for the consent to be signed by a family member in a specific order.
The requirement for informed consent can be waived if there is no ability to get the consent of the subject (e.g., the subject is incompetent) and there is not enough time to get consent from a legally authorized representative. In this case, both the investigator AND a physician who is not otherwise participating in the investigation must certify in writing that
- The subject is confronted by a life-threatening situation necessitating the use of the test article;
- Informed consent cannot be obtained because of the inability to communicate with, or obtain legally effective consent from the subject;
- There is not sufficient time to obtain consent from the subject’s legally authorized representative; and
- No alternative method of approved or generally recognized therapy is available that provides an equal or greater likelihood of saving the subject’s life
This pathway provides a mechanism for verifying that the intended use meets criteria for providing documentation of correspondence with FDA, including the emergency IND approval, the expanded access protocol/treatment plan and the proposed informed consent document.
The IRB Chair/Designee, can review and acknowledge the single patient use. This pathway also allows for submission of follow up information on the status of the patient. The acknowledgment of the single patient use by the IRB Chair/Designee, should not be construed as IRB approval. Only proposals that undergo full IRB review can receive IRB approval.
In the event of a waiver of informed consent for an emergency use, the IRB Chair/Designee, will confirm that both the physician holding the emergency IND and a physician who is not otherwise participating in the emergency use have certified in writing all of the following:
- the patient is confronted by a life-threatening situation necessitating use of the test article; informed consent cannot be obtained because of an inability to communicate with, or obtain legally effective consent from, the patient;
- time is not sufficient to obtain consent from the patient’s legal representative;
- no alternative method of approved or generally recognized therapy is available that provides an equal or greater likelihood of saving the patient’s life;
If, in the physician’s opinion, there is not sufficient time to obtain an independent physician’s determination that the criteria are met, the physician holding the emergency IND should make the determination and subsequently obtain (within five working days) a review of his/her determination by a physician not participating in the emergency treatment.
Summary of Submission process for Requesting IRB Concurrence
| Action | Descriptions and Further Information |
|---|---|
| 1. Request Letter of Authorization |
|
| 2. Submit Form 3926 to FDA |
|
| 3. Obtain IRB Concurrence |
|
| 4. Obtain Informed Consent |
|
In addition to the criteria for all expanded access uses listed earlier, the following must also be met:
- The physician must determine that the probable risk to the person from the investigational drug is not greater than the probable risk from the disease or condition; and
- FDA must determine that the patient cannot obtain the drug under another IND or protocol.

UVA Study teams are NOT required to go through CRConnect or Protocol Builder for single patient treatment submission.
If you submit Form FDA 3926 to the FDA and selected the box under the Field 10.b (request for authorization to use alternative IRB review procedures: IRB CONCURRENCE):
Email the following documents via sIRB@virginia.edu : Subject line: Single Patient Treatment Use-IRB Concurrence [Indicate Emergency or Non- Emergency]
- Choose either Emergency Use or Non-Emergency Use for a Single Patient
OR
-
Form 3926 (FDA)-see Instructions for Completion >>>>>>>>>>>>>>>>>>>ANSWER #10b YES
- Letter of Authorization (LOA) from study drug manufacturer (sample template HERE if needed)
- FDA Authorization letter to proceed with single patient treatment
- Single Patient IND# (assigned by FDA)
- Study specific informed consent-See UVA Template
- Treatment Plan/Protocol
- Investigator Brochure (IB)
- If applicable, Contact Investigational Drug Services (IDS): to make them aware of the single patient IND treatment in the event additional precautions need to be in place with IDS. Provide a copy of the IB and protocol/treatment plan
FOLLOW-UP:
After the use of the test article, you are required to notify the UVA IRB-HSR within 5 working days about the status of the patient using the Single Patient Treatment Follow-up form. The FDA and sponsor will need a similar report. Be aware of the reporting requirements noted in the table that follows.
| Action | Timeframe | Descriptions and Further Information |
|---|---|---|
| Safety Reports | As soon as possible |
|
| Amendments | Any time |
|
| Summary | Following completion of treatment |
|
| Annual Report | Within 60 days of the anniversary date |
|
Expanded Access for Intermediate-size populations:
FDA may permit an investigational drug to be used for treatment of a patient population smaller than that typical of a treatment IND or treatment protocol. An intermediate-size population IND has no fixed numerical requirement, but it is for more than one patient and is generally employed when the investigational drug is not actively being developed for marketing. In cases where FDA has received a significant number of requests for individual patient expanded access for the same use, a sponsor may be asked to consolidate expanded access under this category.
In addition to the criteria listed at the beginning of this section for all expanded access, the FDA must also determine that there is enough evidence that the drug is safe at the proposed dose and duration and there is at least preliminary evidence of effectiveness of the drug as a therapeutic option in the patient population. For more information about FDA requirements, please see 21 CFR 312.315.
Procedures for IRB submission of expanded access protocol for intermediate –size populations
Intermediate size expanded access protocols must be submitted through usual procedures through CRConnect and the UVA IRB-HSR and requires full IRB review and approval under FDA regulations. Please see the School of Medicine website for information about preparing an IND submission to FDA. Submit all documents for pre-review via CRConnect and Protocol Builder.
Expanded Access for Large Patient Populations (Treatment IND or Treatment Protocol)
Expanded access protocols for large patient populations are also referred to as treatment IND or treatment protocols. This category is used for widespread treatment use of an investigational drug. A widespread treatment IND is typically used to provide access to a large population and is often used to bridge the gap between completion of clinical trials and marketing approval. In addition to the criteria listed at the beginning of this section for all expanded access, FDA must also determine that the drug is being investigated in a controlled trial under an IND to support a marketing application for the expanded access or all clinical trials of the drug have been completed, the sponsor is actively pursuing marketing for approval of the expanded access and there is sufficient data supporting safety and effectiveness of the drug for the expanded access.
Procedures for IRB submission of expanded access protocols for large patient populations
Expanded access protocols for large patient populations must be submitted per usual procedures through CRConnect and Protocol Builder through the UVA IRB-HSR and requires full IRB review and approval under FDA regulations. Please see the School of Medicine website for information about preparing an IND submission to FDA.
An investigator may withdraw an IND at any time with or without cause. A letter to the FDA is required and copy should be forwarded to the IRB.
The FDA may place an IND on "Clinical Hold" for a number of reasons. Clinical hold means that all study activity is halted pending the FDA-required modifications. When a clinical hold is placed, the IND initiator usually has 30-days to respond back to the FDA.
- A Clinical Hold may be issued if it appears subjects are being exposed to greater risk than had originally been recognized. . Enrollment is halted and subjects currently being treated may only continue on study drug if it is clinically necessary for them to do so. The Clinical Hold is often lifted after adjustments have been made to the study design.
- A Clinical Hold may also be issued if the researcher's qualifications are called into serious question or if the study design proves fatally flawed in such a way that no meaningful data will be gleaned and/or no meaningful results will be determined from the data.
The FDA may also terminate an IND if clear and compelling danger to the research subjects is present or if there is evidence of fraud on the part of the investigator. Termination is usually only undertaken when reactivation is not anticipated.
Investigators as Sponsors
If an investigator is the developer of the drug, biologic or medical device, and no commercial manufacturer is involved, then the investigator is also the sponsor for the purposes of designing and organizing clinical trials.
Sponsors also have important administrative and reporting requirements above and beyond those of investigators. Faculty contemplating the dual role of sponsor-investigator should consult with the School of Medicine Clinical Trials Office (SOM CTO) about the additional responsibilities that entails.
The sponsor must declare any individual financial conflict(s) of interests in the research and develop a management plan that is approved by the University.
Should an investigator associated with the University of Virginia or the University sponsor a multi-site study, that investigator is required to meet all the responsibilities of a sponsor as determined by DHHS guidance.
A common protocol is required for all multi-site trials. See Multi-Site Studies for additional information. At the time of initial review the IRB will require an approval from the SOM CTO who will assess the procedures for dissemination of protocol information (e.g. unanticipated problems involving risks to subjects or others, protocol modifications, interim findings) to all participating sites. In addition, the UVA PI must ensure that investigators at other research sites submit and follow requirements directed by their local IRBs.
IRB policies and procedures from each approving institution will be followed by researchers at that site. All required reports will be provided to the local IRB as per their policy. The coordinating PI at the University of Virginia will be responsible for providing local information as well as unanticipated problems involving risks to subjects or others, protocol modifications, or interim findings that may affect the UVA IRB's continuing approval of the research.
If a UVA faculty member is the principal investigator on an IND or IDE, the IRB-HSR will require an approval from the School of Medicine Clinical Trials Office prior to subjects enrolling in the protocol. Prior to granting approval, the SOM CTO will conduct of review of various items with special focus on areas of FDA interest such as- inclusion/exclusion criteria, safety plan , endpoints, data collection process and the communication plan with other sites , if multi-site. The staff of the SOM CTO will also review Sponsor responsibilities with the PI. Sponsor responsibilities for an IND are found at 21CFR312. Sponsor responsibilities for an IDE are found at 21CFR812